
Research
My research focuses on developing novel statistical machine learning and deep learning models to decode complex, high dimensional, and structured data with a focus in computational biology and genomics. We try to build interpretable models to find the biomarkers that explain the underlying pathophysiology of the disease.
Recent Works
VariantFormer: A Diploid-Aware Transformer for Personalized Gene Expression Prediction
Accurately predicting gene expression from DNA sequence remains a central challenge in human genetics. Current sequence-based models overlook natural genetic variation across individuals, while population-based models are restricted to variants observed within specific cohorts. Here, we present VariantFormer, a 1.2-billion-parameter transformer that predicts gene-level RNA abundance directly from personalized diploid genomes. Trained on 21,004 genome–transcriptome pairs from 2,330 donors, VariantFormer achieves state-of-the-art performance across both sequence- and population-based prediction tasks, while generalizing better to out-of-distribution contexts—including somatic mutation settings in cancer cell lines—and maintaining robustness across ancestries. Beyond expression prediction, VariantFormer improves eQTL effect size estimation compared to prior methods, with notable gains for lower-frequency and ancestry-specific variants. In applications to Alzheimer’s disease, VariantFormer gene embeddings prioritize likely causal genes and relevant tissue contexts, and in silico mutagenesis of known APOE alleles faithfully recovers known risk modifying effects. Together, these results establish VariantFormer as a scalable, diploid-aware framework for variant interpretation and personalized gene expression modeling across tissues and populations.
Motif Driven Structural Variant discovery
Structural Variants (SVs) are large-scale recombination of genomic regions. These variants are often diverse, encompassing simple events like deletion and insertion and complex events like shuffling regions across different chromosomes. SVs usually account for a significant portion of genetic diversity, and they have been found to be key drivers of neuropsychiatric diseases. However, it’s often difficult to discover a high-confidence set of structural variants due to the inability of heuristic-based models to find complex SV signatures.
In this work, we view the problem of SV discovery as a motif-finding problem. Our approach utilizes the graph representation of sequence alignments to find regions of genomic rearrangements. We are developing a learning-based algorithm that can discover motifs as general signatures of SVs. This learning-based method discovers new SVs while providing flexibility over heuristic-based models to identify known SVs.
BEATRICE: Bayesian Fine-mapping from Summary Data using Deep Variational Inference
We introduce a novel framework to identify putative causal variants from GWAS summary statistics. Our approach relies on a hierarchical Bayesian model that imposes a binary concrete prior on the set of causal variants. We derive a variational algorithm for this fine-mapping problem by minimizing the KL divergence between an approximate density and the posterior probability distribution of the causal configurations. We use a binary concrete distribution as our approximation, whose unique properties allow it to be optimized using stochastic gradient descent. Correspondingly, we use a deep neural network as an inference machine to estimate the parameters of our proposal distribution. The stochastic optimization procedure allows us to simultaneously sample from the space of causal configurations. We use these samples to compute the posterior inclusion probabilities and determine credible sets for each causal variant. We conduct a detailed simulation study to quantify the performance of our framework across different numbers of causal variants and different noise paradigms, as defined by the relative genetic contributions of causal and non-causal variants. Finally, we perform a comparative analysis against two state-of-the-art baseline methods for fine-mapping. We demonstrate that our framework achieves uniformly better coverage with comparable power and set sizes. bioRxiv
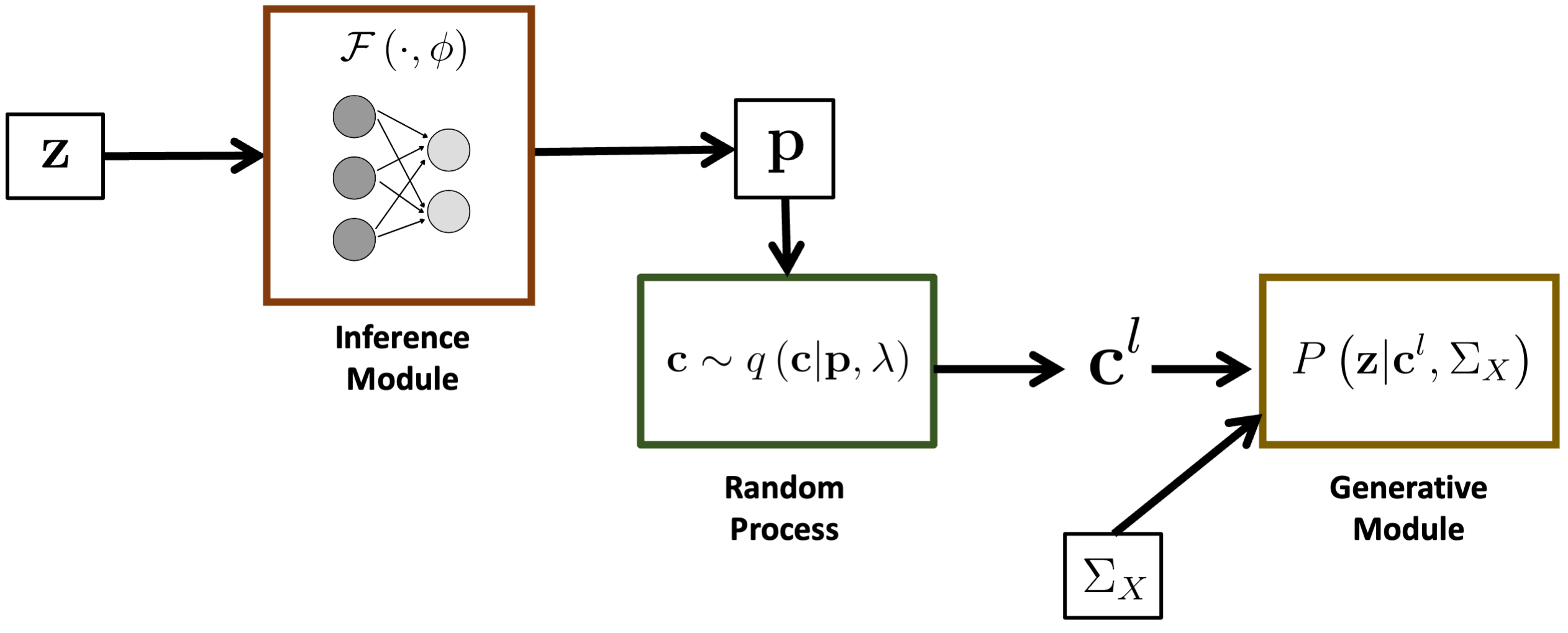
Overview of the BEATRICE framework.
A Biologically Interpretable Graph Convolutional Network to Link Genetic Risk Pathways and Neuroimaging Markers of Disease
We propose a novel end-to-end framework for whole-brain and whole-genome imaging-genetics. Our genetics network uses hierarchical graph convolution and pooling operations to embed subject-level data onto a low-dimensional latent space. The hierarchical network implicitly tracks the convergence of genetic risk across well-established biological pathways, while an attention mechanism automatically identifies the salient edges of this network at the subject level. In parallel, our imaging network projects multimodal data onto a set of latent embeddings. For interpretability, we implement a Bayesian feature selection strategy to extract the discriminative imaging biomarkers; these feature weights are optimized alongside the other model parameters. We couple the imaging and genetic embeddings with a predictor network, to ensure that the learned representations are linked to phenotype. We evaluate our framework on a schizophrenia dataset that includes two functional MRI paradigms and gene scores derived from Single Nucleotide Polymorphism data. Using repeated 10-fold cross-validation, we show that our imaging-genetics fusion achieves the better classification performance than state-of-the-art baselines. In an exploratory analysis, we further show that the biomarkers identified by our model are reproducible and closely associated with deficits in schizophrenia. bioRxiv
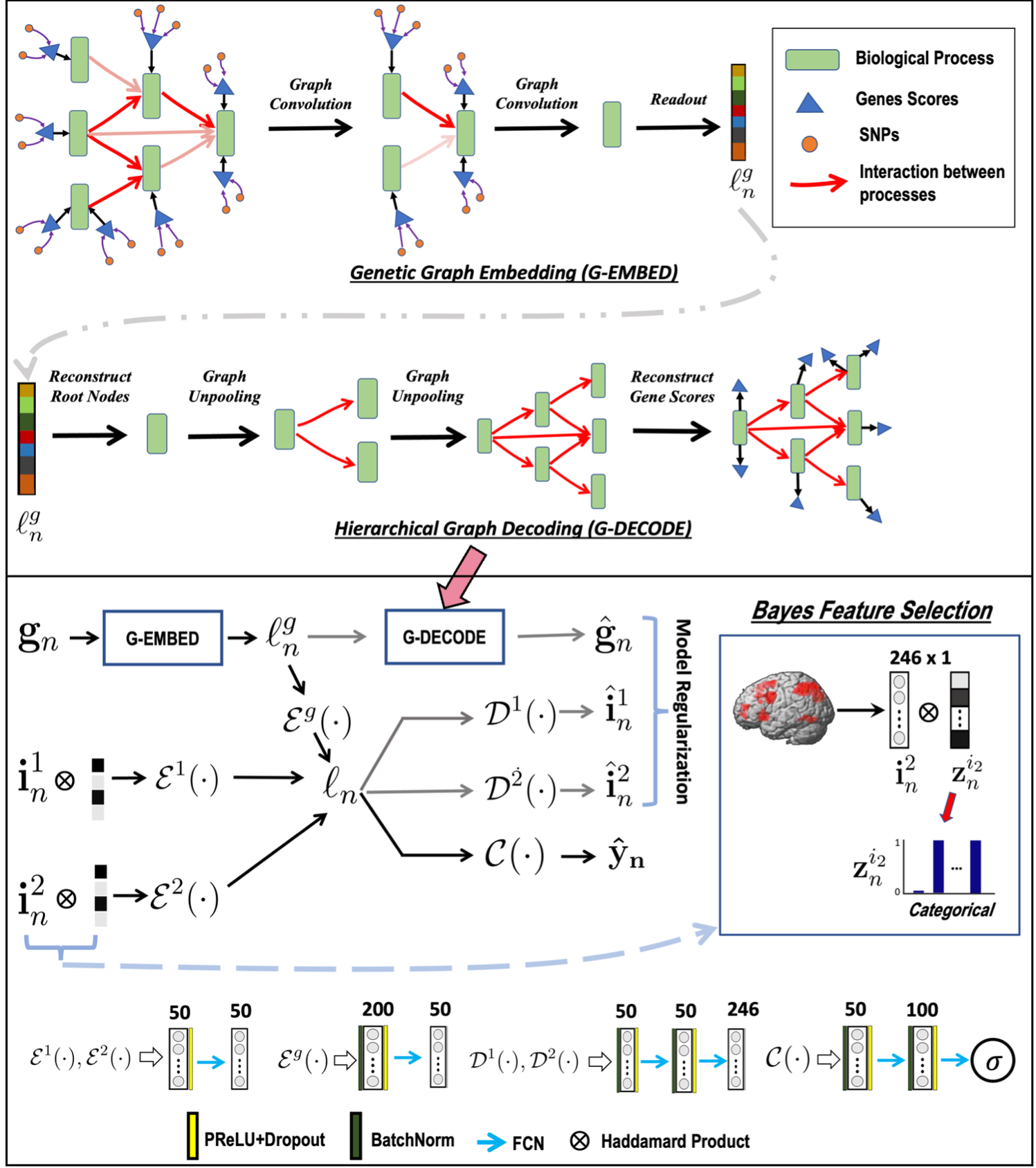
Overview of the GUIDE framework. Top: Gene embedding using attention based hierarchical graph convolution. We also depict the unpooling operation used as a regularizer. Bottom: Imaging and genetics integration; both modalities are coupled for disease classification. The variables $\{\mathbf{i}_n^1, \mathbf{i}_n^2\}$ correspond to the imaging data, and $\mathbf{g}_n$ is the genetic data. $\mathcal{E}(\cdot)$, $\mathcal{D}(\cdot)$, $\mathcal{C}(\cdot)$ are the feature extraction, model regularization, and classification operations, respectively.
G-MIND: An End-to-End Multimodal Imaging-Genetics Framework for Biomarker Identification and Disease Classification
We propose a novel deep neural network architecture to integrate imaging and genetics data, as guided by diagnosis, while preserving interpretability. Our model consists of an encoder, a decoder and a classifier. The encoder learns a non-linear subspace shared between the input data modalities. The classifier and the decoder act as regularizers to ensure that the low-dimensional encoding captures predictive differences between patients and controls. We use a learnable dropout layer to extract interpretable biomarkers from the data, and our unique training strategy can easily accommodate missing data modalities across subjects. We have evaluated our model on a population study of schizophrenia that includes two functional MRI (fMRI) paradigms and Single Nucleotide Polymorphism (SNP) data. Using 10-fold cross validation, we demonstrate that our model achieves better classification accuracy than baseline methods, and that this performance generalizes to a second dataset collected at a different site. The biomarkers identified by our model are closely associated with the well-documented deficits in schizophrenia.
ArXiv
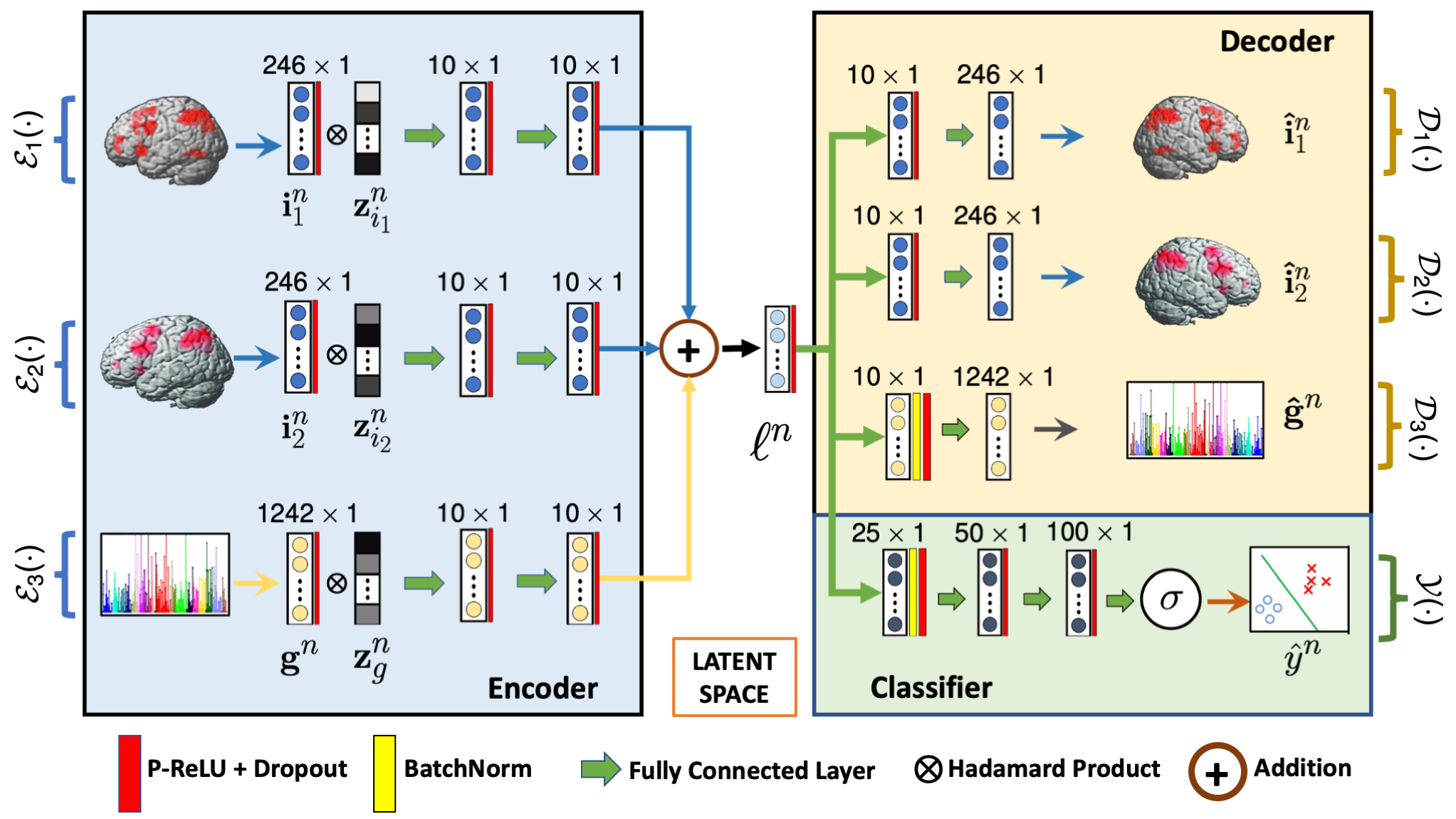
G-MIND architecture. The inputs $\{\mathbf{i}_1, \mathbf{i}_2\}$ and $\{\mathbf{g}\}$ corresponds to the two imaging modalities and genetic data, respectively. $\mathcal{E}_i(\cdot)$ and $\mathcal{D}_i(\cdot)$ captures the encoding and decoding operations, and $\mathcal{Y}(\cdot)$ captures the classification operation. $\mathbf{z}_{i}$ is the learnable dropout mask, and $\mathbf{\ell}^n$ is the low dimensional latent space.
Bridging Imaging, Genetics, and Diagnosis in a Coupled Low-Dimensional Framework
We propose a joint dictionary learning framework that couples imaging and genetics data in a low dimensional subspace as guided by clinical diagnosis. We use a graph regularization penalty to simultaneously capture inter-regional brain interactions and identify the representative set anatomical basis vectors that span the low dimensional space. We further employ group sparsity to find the representative set of genetic basis vectors that span the same latent space. Finally, the latent projection is used to classify patients versus controls. We have evaluated our model on two task fMRI paradigms and single nucleotide polymorphism (SNP) data from schizophrenic patients and matched neurotypical controls. We employ a ten fold cross validation technique to show the predictive power of our model. We compare our model with canonical correlation analysis of imaging and genetics data and random forest classification. Our approach shows better prediction accuracy on both task datasets. Moreover, the implicated brain regions and genetic variants underlie the well documented deficits in schizophrenia.
Link
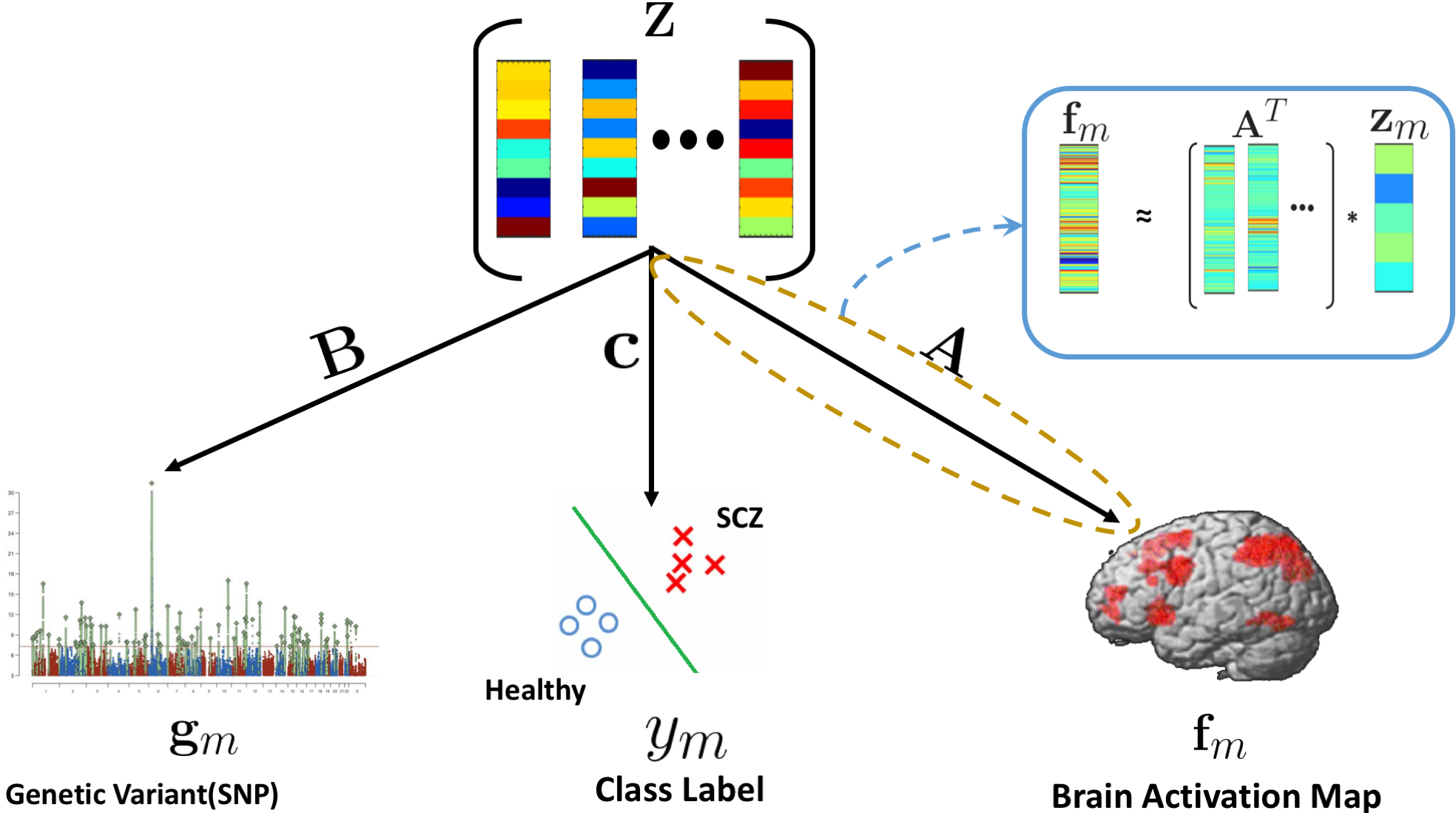
The generative process linking imaging ( $\mathbf{f}_m$), genetics ( $\mathbf{g}_m$), and diagnosis ( $y_m$). The imaging model is shown as a linear projection. The genetics model parallels this, whereas the classification is based on a logistic regression.
A generative-predictive framework to capture altered brain activity in fMRI and its association with genetic risk: application to Schizophrenia
We present a generative-predictive framework that captures the differences in regional brain activity between a neurotypical cohort and a clinical population, as guided by patient-specific genetic risk. Our model assumes that the functional activations in the neurotypical subjects are distributed around a population mean, and that the altered brain activity in neuropsychiatric patients is defined via deviations from this neurotypical mean. We employ group sparsity to identify a set of brain regions that simultaneously explain the salient functional differences and specify a set of basis vector, that span the low dimensional data subspace. The patient-specific projections onto this subspace are used as feature vectors to identify multivariate associations with genetic risk. We have evaluated our model on a task-based fMRI dataset from a population study of schizophrenia. We compare our model with two baseline methods, regression using Least Absolute Shrinkage and Selection Operator (LASSO) and Random Forest (RF) regression, which establishes direct association between the brain activity during a working memory task and schizophrenia polygenic risk. Our model demonstrates greater consistency and robustness across bootstrapping experiments than the machine learning baselines. Moreover, the set of brain regions implicated by our model underlie the well documented executive cognitive deficits in schizophrenia.
Link
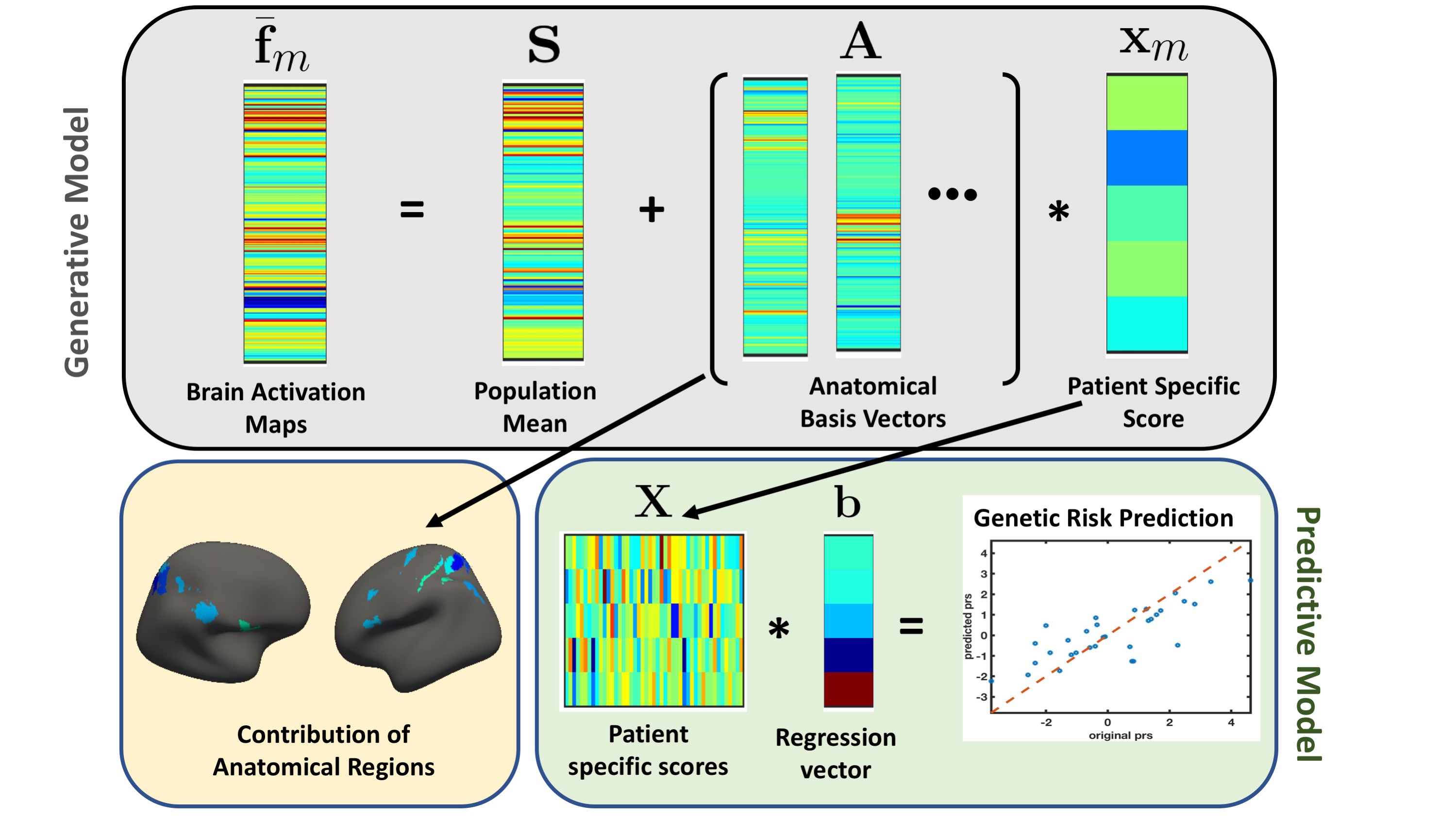
The joint modelling framework to capture brain activity and genetic risk. The gray box represents the generative part of the model for a single schizophrenia patient. We captured altered brain activity in the patients as deviations from the population mean. The major contribution of the anatomical regions to overall deviation are shown as surface plots in the yellow box. The green box is the predictive part of the model that track the genetic risk as linear regression.
Past Research
Deep Deformable Image Registration
Deformable registration is ubiquitous in medical image analysis. Many deformable registration methods minimize sum of squared difference (SSD) as the registration cost with respect to deformable model parameters. In this work, we construct a tight upper bound of the SSD registration cost by using a fully convolutional neural network (FCNN) in the registration pipeline. The upper bound SSD (UB-SSD) enhances the original deformable model parameter space by adding a heatmap output from FCNN. Next, we minimize this UB-SSD by adjusting both the parameters of the FCNN and the parameters of the deformable model in coordinate descent. Our coordinate descent framework is end-to-end and it can work with any deformable registration method that uses SSD. We demonstrate experimentally that our method enhances the accuracy of deformable registration algorithms significantly on two publicly available 3D brain MRI data sets.
Link
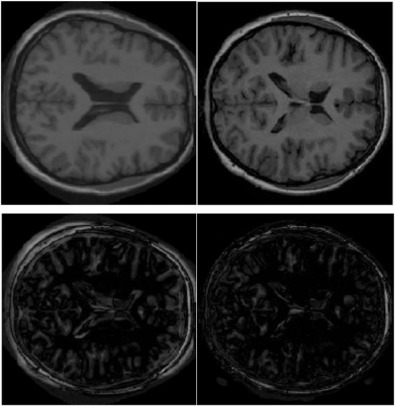
Top left: A moving image slice; top right: a fixed image slice; bottom left: residual image from demons. bottom right: residual using DDR+demons.
A novel non-rigid registration algorithm for zebrafish larval images
Precise three-dimensional mapping of a large number of gene expression patterns, neuronal types and connections to an anatomical reference helps us to understand the vertebrate brain and its development. Zebrafish has evolved as a model organism for such study. In this paper, we propose a novel non-rigid registration algorithm for volumetric zebrafish larval image datasets. A coarse affine registration using the L-BFGS algorithm is applied first on the moving dataset. We then divide this coarsely registered moving image and the reference image into a union of overlapping patches. Minimum weight bipartite graph matching algorithm is employed to find the correspondence between the two sets of patches. The corresponding patches are then registered using the diffeomorphic demons method with proper intra-patch regularization. For each voxel lying in the overlapping regions, we impose inter-patch regularization through a composite transformation obtained from the adjacent transformation fields. Experimental results on four multi-view confocal 3D datasets show the advantage of the proposed solution over the existing ViBE-Z software.
Link
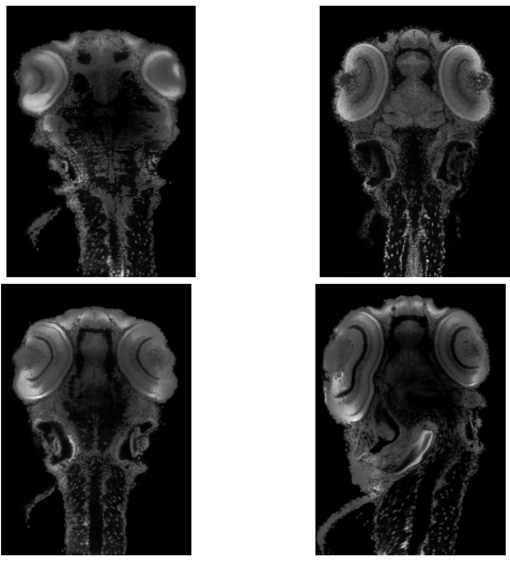
Top Left: Moving Image, Top Right: Fixed Image, Bottom Left: Registration using our method, Bottom Right: Registration using VibeZ. All implementations are done in 3D and all results are shown for the same 2D slice of dataset 2.